API Reference
This part of the documentation details the complete BioSPPy API.
Packages
Modules
biosppy.biometrics
This module provides classifier interfaces for identity recognition (biometrics) applications. The core API methods are: * enroll: add a new subject; * dismiss: remove an existing subject; * identify: determine the identity of collected biometric dataset; * authenticate: verify the identity of collected biometric dataset.
- copyright:
2015-2018 by Instituto de Telecomunicacoes
- license:
BSD 3-clause, see LICENSE for more details.
- class biosppy.biometrics.BaseClassifier[source]
Bases:
objectBase biometric classifier class.
This class is a skeleton for actual classifier classes. The following methods must be overridden or adapted to build a new classifier:
__init__
_authenticate
_get_thresholds
_identify
_prepare
_train
_update
- EER_IDX
Reference index for the Equal Error Rate.
- Type:
int
- EER_IDX = 0
- authenticate(data, subject, threshold=None)[source]
Authenticate a set of feature vectors, allegedly belonging to the given subject.
- Parameters:
data (array) – Input test data.
subject (hashable) – Subject identity.
threshold (int, float, optional) – Authentication threshold.
- Returns:
decision (array) – Authentication decision for each input sample.
- batch_train(data=None)[source]
Train the classifier in batch mode.
- Parameters:
data (dict) – Dictionary holding training data for each subject; if the object for a subject is None, performs a dismiss.
- check_subject(subject)[source]
Check if a subject is enrolled.
- Parameters:
subject (hashable) – Subject identity.
- Returns:
check (bool) – If True, the subject is enrolled.
- classmethod cross_validation(data, labels, cv, thresholds=None, **kwargs)[source]
Perform Cross Validation (CV) on a data set.
- Parameters:
data (array) – An m by n array of m data samples in an n-dimensional space.
labels (list, array) – A list of m class labels.
cv (CV iterator) – A sklearn.model_selection iterator.
thresholds (array, optional) – Classifier thresholds to use.
**kwargs (dict, optional) – Classifier parameters.
- Returns:
runs (list) – Evaluation results for each CV run.
assessment (dict) – Final CV biometric statistics.
- dismiss(subject=None, deferred=False)[source]
Remove a subject.
- Parameters:
subject (hashable) – Subject identity.
deferred (bool, optional) – If True, computations are delayed until flush is called.
- Raises:
SubjectError – If the subject to remove is not enrolled.
Notes
When using deferred calls, a dismiss overrides a previous enroll for the same subject.
- enroll(data=None, subject=None, deferred=False)[source]
Enroll new data for a subject.
If the subject is already enrolled, new data is combined with existing data.
- Parameters:
data (array) – Data to enroll.
subject (hashable) – Subject identity.
deferred (bool, optional) – If True, computations are delayed until flush is called.
Notes
When using deferred calls, an enroll overrides a previous dismiss for the same subject.
- evaluate(data, thresholds=None, path=None, show=False)[source]
Assess the performance of the classifier in both authentication and identification scenarios.
- Parameters:
data (dict) – Dictionary holding test data for each subject.
thresholds (array, optional) – Classifier thresholds to use.
path (str, optional) – If provided, the plot will be saved to the specified file.
show (bool, optional) – If True, show a summary plot.
- Returns:
classification (dict) – Classification results.
assessment (dict) – Biometric statistics.
- get_auth_thr(subject, ready=False)[source]
Get the authentication threshold of a subject.
- Parameters:
subject (hashable) – Subject identity.
ready (bool, optional) – If True, subject is the internal classifier label.
- Returns:
threshold (int, float) – Threshold value.
- get_id_thr(subject, ready=False)[source]
Get the identification threshold of a subject.
- Parameters:
subject (hashable) – Subject identity.
ready (bool, optional) – If True, subject is the internal classifier label.
- Returns:
threshold (int, float) – Threshold value.
- get_thresholds(force=False)[source]
Get an array of reasonable thresholds.
- Parameters:
force (bool, optional) – If True, forces generation of thresholds.
- Returns:
ths (array) – Generated thresholds.
- identify(data, threshold=None)[source]
Identify a set of feature vectors.
- Parameters:
data (array) – Input test data.
threshold (int, float, optional) – Identification threshold.
- Returns:
subjects (list) – Identity of each input sample.
- io_del(label)[source]
Delete subject data.
- Parameters:
label (str) – Internal classifier subject label.
- io_load(label)[source]
Load enrolled subject data.
- Parameters:
label (str) – Internal classifier subject label.
- Returns:
data (array) – Subject data.
- io_save(label, data)[source]
Save subject data.
- Parameters:
label (str) – Internal classifier subject label.
data (array) – Subject data.
- list_subjects()[source]
List all the enrolled subjects.
- Returns:
subjects (list) – Enrolled subjects.
- classmethod load(path)[source]
Load classifier instance from a file.
- Parameters:
path (str) – Source file path.
- Returns:
clf (object) – Loaded classifier instance.
- save(path)[source]
Save classifier instance to a file.
- Parameters:
path (str) – Destination file path.
- set_auth_thr(subject, threshold, ready=False)[source]
Set the authentication threshold of a subject.
- Parameters:
subject (hashable) – Subject identity.
threshold (int, float) – Threshold value.
ready (bool, optional) – If True, subject is the internal classifier label.
- exception biosppy.biometrics.CombinationError[source]
Bases:
ExceptionException raised when the combination method fails.
- class biosppy.biometrics.KNN(k=3, metric='euclidean', metric_args=None)[source]
Bases:
BaseClassifierK Nearest Neighbors (k-NN) biometric classifier.
- Parameters:
k (int, optional) – Number of neighbors.
metric (str, optional) – Distance metric.
metric_args (dict, optional) – Additional keyword arguments are passed to the distance function.
- EER_IDX
Reference index for the Equal Error Rate.
- Type:
int
- EER_IDX = 0
- class biosppy.biometrics.SVM(C=1.0, kernel='linear', degree=3, gamma='auto', coef0=0.0, shrinking=True, tol=0.001, cache_size=200, max_iter=-1, random_state=None)[source]
Bases:
BaseClassifierSupport Vector Machines (SVM) biometric classifier.
Wraps the ‘OneClassSVM’ and ‘SVC’ classes from ‘scikit-learn’.
- Parameters:
C (float, optional) – Penalty parameter C of the error term.
kernel (str, optional) – Specifies the kernel type to be used in the algorithm. It must be one of ‘linear’, ‘poly’, ‘rbf’, ‘sigmoid’, ‘precomputed’ or a callable. If none is given, ‘rbf’ will be used. If a callable is given it is used to precompute the kernel matrix.
degree (int, optional) – Degree of the polynomial kernel function (‘poly’). Ignored by all other kernels.
gamma (float, optional) – Kernel coefficient for ‘rbf’, ‘poly’ and ‘sigmoid’. If gamma is ‘auto’ then 1/n_features will be used instead.
coef0 (float, optional) – Independent term in kernel function. It is only significant in ‘poly’ and ‘sigmoid’.
shrinking (bool, optional) – Whether to use the shrinking heuristic.
tol (float, optional) – Tolerance for stopping criterion.
cache_size (float, optional) – Specify the size of the kernel cache (in MB).
max_iter (int, optional) – Hard limit on iterations within solver, or -1 for no limit.
random_state (int, RandomState, optional) – The seed of the pseudo random number generator to use when shuffling the data for probability estimation.
- EER_IDX
Reference index for the Equal Error Rate.
- Type:
int
- EER_IDX = -1
- exception biosppy.biometrics.SubjectError(subject=None)[source]
Bases:
ExceptionException raised when the subject is unknown.
- exception biosppy.biometrics.UntrainedError[source]
Bases:
ExceptionException raised when classifier is not trained.
- biosppy.biometrics.assess_classification(results=None, thresholds=None)[source]
Assess the performance of a biometric classification test.
- Parameters:
results (dict) – Classification results.
thresholds (array) – Classifier thresholds.
- Returns:
assessment (dict) – Classification assessment.
- biosppy.biometrics.assess_runs(results=None, subjects=None)[source]
Assess the performance of multiple biometric classification runs.
- Parameters:
results (list) – Classification assessment for each run.
subjects (list) – Common target subject classes.
- Returns:
assessment (dict) – Global classification assessment.
- biosppy.biometrics.combination(results=None, weights=None)[source]
Combine results from multiple classifiers.
- Parameters:
results (dict) – Results for each classifier.
weights (dict, optional) – Weight for each classifier.
- Returns:
decision (object) – Consensus decision.
confidence (float) – Confidence estimate of the decision.
counts (array) – Weight for each possible decision outcome.
classes (array) – List of possible decision outcomes.
- biosppy.biometrics.cross_validation(labels, n_iter=10, test_size=0.1, train_size=None, random_state=None)[source]
Return a Cross Validation (CV) iterator.
Wraps the StratifiedShuffleSplit iterator from sklearn.model_selection. This iterator returns stratified randomized folds, which preserve the percentage of samples for each class.
- Parameters:
labels (list, array) – List of class labels for each data sample.
n_iter (int, optional) – Number of splitting iterations.
test_size (float, int, optional) – If float, represents the proportion of the dataset to include in the test split; if int, represents the absolute number of test samples.
train_size (float, int, optional) – If float, represents the proportion of the dataset to include in the train split; if int, represents the absolute number of train samples.
random_state (int, RandomState, optional) – The seed of the pseudo random number generator to use when shuffling the data.
- Returns:
cv (CV iterator) – Cross Validation iterator.
- biosppy.biometrics.get_auth_rates(TP=None, FP=None, TN=None, FN=None, thresholds=None)[source]
Compute authentication rates from the confusion matrix.
- Parameters:
TP (array) – True Positive counts for each classifier threshold.
FP (array) – False Positive counts for each classifier threshold.
TN (array) – True Negative counts for each classifier threshold.
FN (array) – False Negative counts for each classifier threshold.
thresholds (array) – Classifier thresholds.
- Returns:
Acc (array) – Accuracy at each classifier threshold.
TAR (array) – True Accept Rate at each classifier threshold.
FAR (array) – False Accept Rate at each classifier threshold.
FRR (array) – False Reject Rate at each classifier threshold.
TRR (array) – True Reject Rate at each classifier threshold.
EER (array) – Equal Error Rate points, with format (threshold, rate).
Err (array) – Error rate at each classifier threshold.
PPV (array) – Positive Predictive Value at each classifier threshold.
FDR (array) – False Discovery Rate at each classifier threshold.
NPV (array) – Negative Predictive Value at each classifier threshold.
FOR (array) – False Omission Rate at each classifier threshold.
MCC (array) – Matthrews Correlation Coefficient at each classifier threshold.
- biosppy.biometrics.get_id_rates(H=None, M=None, R=None, N=None, thresholds=None)[source]
Compute identification rates from the confusion matrix.
- Parameters:
H (array) – Hit counts for each classifier threshold.
M (array) – Miss counts for each classifier threshold.
R (array) – Reject counts for each classifier threshold.
N (int) – Number of test samples.
thresholds (array) – Classifier thresholds.
- Returns:
Acc (array) – Accuracy at each classifier threshold.
Err (array) – Error rate at each classifier threshold.
MR (array) – Miss Rate at each classifier threshold.
RR (array) – Reject Rate at each classifier threshold.
EID (array) – Error of Identification points, with format (threshold, rate).
EER (array) – Equal Error Rate points, with format (threshold, rate).
- biosppy.biometrics.get_subject_results(results=None, subject=None, thresholds=None, subjects=None, subject_dict=None, subject_idx=None)[source]
Compute authentication and identification performance metrics for a given subject.
- Parameters:
results (dict) – Classification results.
subject (hashable) – True subject label.
thresholds (array) – Classifier thresholds.
subjects (list) – Target subject classes.
subject_dict (bidict) – Subject-label conversion dictionary.
subject_idx (list) – Subject index.
- Returns:
assessment (dict) – Authentication and identification results.
- biosppy.biometrics.majority_rule(labels=None, random=True)[source]
Determine the most frequent class label.
- Parameters:
labels (array, list) – List of clas labels.
random (bool, optional) – If True, will choose randomly in case of tied classes, otherwise the first element is chosen.
- Returns:
decision (object) – Consensus decision.
count (int) – Number of elements of the consensus decision.
biosppy.clustering
This module provides various unsupervised machine learning (clustering) algorithms.
- copyright:
2015-2018 by Instituto de Telecomunicacoes
- license:
BSD 3-clause, see LICENSE for more details.
- biosppy.clustering.centroid_templates(data=None, clusters=None, ntemplates=1)[source]
Template selection based on cluster centroids.
- Parameters:
data (array) – An m by n array of m data samples in an n-dimensional space.
clusters (dict) – Dictionary with the sample indices (rows from ‘data’) for each cluster.
ntemplates (int, optional) – Number of templates to extract; if more than 1, k-means is used to obtain more templates.
- Returns:
templates (array) – Selected templates from the input data.
- biosppy.clustering.coassoc_partition(coassoc=None, k=0, linkage='average')[source]
Extract the consensus partition from a co-association matrix using hierarchical agglomerative methods.
- Parameters:
coassoc (array) – Co-association matrix.
k (int, optional) – Number of clusters to extract; if 0 uses the life-time criterion.
linkage (str, optional) – Linkage criterion for final partition extraction; one of ‘average’, ‘complete’, ‘single’, or ‘weighted’.
- Returns:
clusters (dict) – Dictionary with the sample indices (rows from ‘data’) for each found cluster; outliers have key -1; clusters are assigned integer keys starting at 0.
- biosppy.clustering.consensus(data=None, k=0, linkage='average', fcn=None, grid=None)[source]
Perform clustering based in an ensemble of partitions.
- Parameters:
data (array) – An m by n array of m data samples in an n-dimensional space.
k (int, optional) – Number of clusters to extract; if 0 uses the life-time criterion.
linkage (str, optional) – Linkage criterion for final partition extraction; one of ‘average’, ‘centroid’, ‘complete’, ‘median’, ‘single’, ‘ward’, or ‘weighted’.
fcn (function) – A clustering function.
grid (dict, list, optional) – A (list of) dictionary with parameters for each run of the clustering method (see sklearn.model_selection.ParameterGrid).
- Returns:
clusters (dict) – Dictionary with the sample indices (rows from ‘data’) for each found cluster; outliers have key -1; clusters are assigned integer keys starting at 0.
- biosppy.clustering.consensus_kmeans(data=None, k=0, linkage='average', nensemble=100, kmin=None, kmax=None)[source]
Perform clustering based on an ensemble of k-means partitions.
- Parameters:
data (array) – An m by n array of m data samples in an n-dimensional space.
k (int, optional) – Number of clusters to extract; if 0 uses the life-time criterion.
linkage (str, optional) – Linkage criterion for final partition extraction; one of ‘average’, ‘centroid’, ‘complete’, ‘median’, ‘single’, ‘ward’, or ‘weighted’.
nensemble (int, optional) – Number of partitions in the ensemble.
kmin (int, optional) – Minimum k for the k-means partitions; defaults to
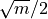 .
.kmax (int, optional) – Maximum k for the k-means partitions; defaults to
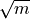 .
.
- Returns:
clusters (dict) – Dictionary with the sample indices (rows from ‘data’) for each found cluster; outliers have key -1; clusters are assigned integer keys starting at 0.
- biosppy.clustering.create_coassoc(ensemble=None, N=None)[source]
Create the co-association matrix from a clustering ensemble.
- Parameters:
ensemble (list) – Clustering ensemble partitions.
N (int) – Number of data samples.
- Returns:
coassoc (array) – Co-association matrix.
- biosppy.clustering.create_ensemble(data=None, fcn=None, grid=None)[source]
Create an ensemble of partitions of the data using the given clustering method.
- Parameters:
data (array) – An m by n array of m data samples in an n-dimensional space.
fcn (function) – A clustering function.
grid (dict, list, optional) – A (list of) dictionary with parameters for each run of the clustering method (see sklearn.model_selection.ParameterGrid).
- Returns:
ensemble (list) – Obtained ensemble partitions.
- biosppy.clustering.dbscan(data=None, min_samples=5, eps=0.5, metric='euclidean', metric_args=None)[source]
Perform clustering using the DBSCAN algorithm [EKSX96].
The algorithm works by grouping data points that are closely packed together (with many nearby neighbors), marking as outliers points that lie in low-density regions.
- Parameters:
data (array) – An m by n array of m data samples in an n-dimensional space.
min_samples (int, optional) – Minimum number of samples in a cluster.
eps (float, optional) – Maximum distance between two samples in the same cluster.
metric (str, optional) – Distance metric (see scipy.spatial.distance).
metric_args (dict, optional) – Additional keyword arguments to pass to the distance function.
- Returns:
clusters (dict) – Dictionary with the sample indices (rows from ‘data’) for each found cluster; outliers have key -1; clusters are assigned integer keys starting at 0.
References
[EKSX96]M. Ester, H. P. Kriegel, J. Sander, and X. Xu, “A Density-Based Algorithm for Discovering Clusters in Large Spatial Databases with Noise”, Proceedings of the 2nd International Conf. on Knowledge Discovery and Data Mining, pp. 226-231, 1996.
- biosppy.clustering.hierarchical(data=None, k=0, linkage='average', metric='euclidean', metric_args=None)[source]
Perform clustering using hierarchical agglomerative algorithms.
- Parameters:
data (array) – An m by n array of m data samples in an n-dimensional space.
k (int, optional) – Number of clusters to extract; if 0 uses the life-time criterion.
linkage (str, optional) – Linkage criterion; one of ‘average’, ‘centroid’, ‘complete’, ‘median’, ‘single’, ‘ward’, or ‘weighted’.
metric (str, optional) – Distance metric (see ‘biosppy.metrics’).
metric_args (dict, optional) – Additional keyword arguments to pass to the distance function.
- Returns:
clusters (dict) – Dictionary with the sample indices (rows from ‘data’) for each found cluster; outliers have key -1; clusters are assigned integer keys starting at 0.
- Raises:
TypeError – If ‘metric’ is not a string.
ValueError – When the ‘linkage’ is unknown.
ValueError – When ‘metric’ is not ‘euclidean’ when using ‘centroid’, ‘median’, or ‘ward’ linkage.
ValueError – When ‘k’ is larger than the number of data samples.
- biosppy.clustering.kmeans(data=None, k=None, init='random', max_iter=300, n_init=10, tol=0.0001)[source]
Perform clustering using the k-means algorithm.
- Parameters:
data (array) – An m by n array of m data samples in an n-dimensional space.
k (int) – Number of clusters to extract.
init (str, array, optional) – If string, one of ‘random’ or ‘k-means++’; if array, it should be of shape (n_clusters, n_features), specifying the initial centers.
max_iter (int, optional) – Maximum number of iterations.
n_init (int, optional) – Number of initializations.
tol (float, optional) – Relative tolerance to declare convergence.
- Returns:
clusters (dict) – Dictionary with the sample indices (rows from ‘data’) for each found cluster; outliers have key -1; clusters are assigned integer keys starting at 0.
- biosppy.clustering.mdist_templates(data=None, clusters=None, ntemplates=1, metric='euclidean', metric_args=None)[source]
Template selection based on the MDIST method [UlRJ04].
Extends the original method with the option of also providing a data clustering, in which case the MDIST criterion is applied for each cluster [LCSF14].
- Parameters:
data (array) – An m by n array of m data samples in an n-dimensional space.
clusters (dict, optional) – Dictionary with the sample indices (rows from data) for each cluster.
ntemplates (int, optional) – Number of templates to extract.
metric (str, optional) – Distance metric (see scipy.spatial.distance).
metric_args (dict, optional) – Additional keyword arguments to pass to the distance function.
- Returns:
templates (array) – Selected templates from the input data.
References
[UlRJ04]U. Uludag, A. Ross, A. Jain, “Biometric template selection and update: a case study in fingerprints”, Pattern Recognition 37, 2004
[LCSF14]A. Lourenco, C. Carreiras, H. Silva, A. Fred, “ECG biometrics: A template selection approach”, 2014 IEEE International Symposium on Medical Measurements and Applications (MeMeA), 2014
- biosppy.clustering.outliers_dbscan(data=None, min_samples=5, eps=0.5, metric='euclidean', metric_args=None)[source]
Perform outlier removal using the DBSCAN algorithm.
- Parameters:
data (array) – An m by n array of m data samples in an n-dimensional space.
min_samples (int, optional) – Minimum number of samples in a cluster.
eps (float, optional) – Maximum distance between two samples in the same cluster.
metric (str, optional) – Distance metric (see scipy.spatial.distance).
metric_args (dict, optional) – Additional keyword arguments to pass to the distance function.
- Returns:
clusters (dict) – Dictionary with the sample indices (rows from ‘data’) for the outliers (key -1) and the normal (key 0) groups.
templates (dict) – Elements from ‘data’ for the outliers (key -1) and the normal (key 0) groups.
- biosppy.clustering.outliers_dmean(data=None, alpha=0.5, beta=1.5, metric='euclidean', metric_args=None, max_idx=None)[source]
Perform outlier removal using the DMEAN algorithm [LCSF13].
- A sample is considered valid if it cumulatively verifies:
distance to average template smaller than a (data derived) threshold ‘T’;
sample minimum greater than a (data derived) threshold ‘M’;
sample maximum smaller than a (data derived) threshold ‘N’;
position of the sample maximum is the same as the given index [optional].
For a set of

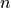 samples:
samples: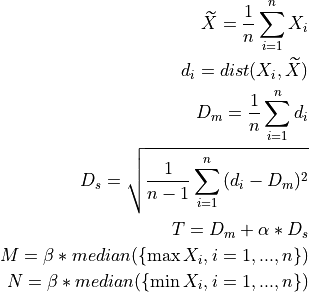
- Parameters:
data (array) – An m by n array of m data samples in an n-dimensional space.
alpha (float, optional) – Parameter for the distance threshold.
beta (float, optional) – Parameter for the maximum and minimum thresholds.
metric (str, optional) – Distance metric (see scipy.spatial.distance).
metric_args (dict, optional) – Additional keyword arguments to pass to the distance function.
max_idx (int, optional) – Index of the expected maximum.
- Returns:
clusters (dict) – Dictionary with the sample indices (rows from ‘data’) for the outliers (key -1) and the normal (key 0) groups.
templates (dict) – Elements from ‘data’ for the outliers (key -1) and the normal (key 0) groups.
References
[LCSF13]A. Lourenco, H. Silva, C. Carreiras, A. Fred, “Outlier Detection in Non-intrusive ECG Biometric System”, Image Analysis and Recognition, vol. 7950, pp. 43-52, 2013
biosppy.metrics
This module provides pairwise distance computation methods.
- copyright:
2015-2018 by Instituto de Telecomunicacoes
- license:
BSD 3-clause, see LICENSE for more details.
- biosppy.metrics.cdist(XA, XB, metric='euclidean', **kwargs)[source]
Computes distance between each pair of the two collections of inputs.
Wraps scipy.spatial.distance.cdist.
- Parameters:
XA (array) – An
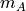 by array of original observations
in an -dimensional space.
by array of original observations
in an -dimensional space.XB (array) – An
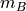 by array of original observations
in an -dimensional space.
by array of original observations
in an -dimensional space.metric (str, function, optional) – The distance metric to use; the distance can be ‘braycurtis’, ‘canberra’, ‘chebyshev’, ‘cityblock’, ‘correlation’, ‘cosine’, ‘dice’, ‘euclidean’, ‘hamming’, ‘jaccard’, ‘kulsinski’, ‘mahalanobis’, ‘matching’, ‘minkowski’, ‘pcosine’, ‘rogerstanimoto’, ‘russellrao’, ‘seuclidean’, ‘sokalmichener’, ‘sokalsneath’, ‘sqeuclidean’, ‘yule’.
kwargs (Possible) –
- pfloat
The p-norm to apply (for Minkowski, weighted and unweighted).
- warray
The weight vector (for weighted Minkowski).
- Varray
The variance vector (for standardized Euclidean).
- VIarray
The inverse of the covariance matrix (for Mahalanobis).
- Returns:
Y (array) – An
by distance matrix is returned. For each
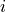 and
and 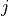 , the metric
, the metric dist(u=XA[i], v=XB[j])is computed and stored in the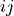 th entry.
th entry.
- biosppy.metrics.pcosine(u, v)[source]
Computes the Cosine distance (positive space) between 1-D arrays.
The Cosine distance (positive space) between u and v is defined as
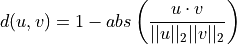
where
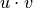 is the dot product of
is the dot product of 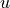 and
and  .
.- Parameters:
u (array) – Input array.
v (array) – Input array.
- Returns:
cosine (float) – Cosine distance between u and v.
- biosppy.metrics.pdist(X, metric='euclidean', **kwargs)[source]
Pairwise distances between observations in n-dimensional space.
Wraps scipy.spatial.distance.pdist.
- Parameters:
X (array) – An m by n array of m original observations in an n-dimensional space.
metric (str, function, optional) – The distance metric to use; the distance can be ‘braycurtis’, ‘canberra’, ‘chebyshev’, ‘cityblock’, ‘correlation’, ‘cosine’, ‘dice’, ‘euclidean’, ‘hamming’, ‘jaccard’, ‘kulsinski’, ‘mahalanobis’, ‘matching’, ‘minkowski’, ‘pcosine’, ‘rogerstanimoto’, ‘russellrao’, ‘seuclidean’, ‘sokalmichener’, ‘sokalsneath’, ‘sqeuclidean’, ‘yule’.
kwargs (Possible) –
- pfloat
The p-norm to apply (for Minkowski, weighted and unweighted).
- warray
The weight vector (for weighted Minkowski).
- Varray
The variance vector (for standardized Euclidean).
- VIarray
The inverse of the covariance matrix (for Mahalanobis).
- Returns:
Y (array) – Returns a condensed distance matrix Y. For each
and
(where 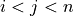 ), the metric
), the metric dist(u=X[i], v=X[j])is computed and stored in entryij.
- biosppy.metrics.squareform(X, force='no', checks=True)[source]
Converts a vector-form distance vector to a square-form distance matrix, and vice-versa.
Wraps scipy.spatial.distance.squareform.
- Parameters:
X (array) – Either a condensed or redundant distance matrix.
force (str, optional) – As with MATLAB(TM), if force is equal to ‘tovector’ or ‘tomatrix’, the input will be treated as a distance matrix or distance vector respectively.
checks (bool, optional) – If checks is set to False, no checks will be made for matrix symmetry nor zero diagonals. This is useful if it is known that
X - X.T1is small anddiag(X)is close to zero. These values are ignored any way so they do not disrupt the squareform transformation.
- Returns:
Y (array) – If a condensed distance matrix is passed, a redundant one is returned, or if a redundant one is passed, a condensed distance matrix is returned.
biosppy.plotting
This module provides utilities to plot data.
- copyright:
2015-2023 by Instituto de Telecomunicacoes
- license:
BSD 3-clause, see LICENSE for more details.
- biosppy.plotting.color_palette(idx)[source]
Color palette to use throughout the biosppy package
- Parameters:
idx (str or int) – identifier of color to use
- Returns:
color_id (str) – hexadecimal color code chosen
- biosppy.plotting.plot_abp(ts=None, raw=None, filtered=None, onsets=None, heart_rate_ts=None, heart_rate=None, path=None, show=False)[source]
Create a summary plot from the output of signals.abp.abp.
- Parameters:
ts (array) – Signal time axis reference (seconds).
raw (array) – Raw ABP signal.
filtered (array) – Filtered ABP signal.
onsets (array) – Indices of ABP pulse onsets.
heart_rate_ts (array) – Heart rate time axis reference (seconds).
heart_rate (array) – Instantaneous heart rate (bpm).
path (str, optional) – If provided, the plot will be saved to the specified file.
show (bool, optional) – If True, show the plot immediately.
- biosppy.plotting.plot_acc(ts=None, raw=None, vm=None, sm=None, units=None, path=None, show=False)[source]
Create a summary plot from the output of signals.acc.acc.
- Parameters:
ts (array) – Signal time axis reference (seconds).
raw (array) – Raw ACC signal.
vm (array) – Vector Magnitude feature of the signal.
sm (array) – Signal Magnitude feature of the signal
units (str, optional) – Units of the vertical axis. If provided, the plot title will include the units information. Default is None.
path (str, optional) – If provided, the plot will be saved to the specified file.
show (bool, optional) – If True, show the plot immediately.
- biosppy.plotting.plot_bcg(ts=None, raw=None, filtered=None, jpeaks=None, templates_ts=None, templates=None, heart_rate_ts=None, heart_rate=None, path=None, show=False)[source]
Create a summary plot from the output of signals.bcg.bcg.
- Parameters:
ts (array) – Signal time axis reference (seconds).
raw (array) – Raw ECG signal.
filtered (array) – Filtered ECG signal.
ipeaks (array) – I-peak location indices.
templates_ts (array) – Templates time axis reference (seconds).
templates (array) – Extracted heartbeat templates.
heart_rate_ts (array) – Heart rate time axis reference (seconds).
heart_rate (array) – Instantaneous heart rate (bpm).
path (str, optional) – If provided, the plot will be saved to the specified file.
show (bool, optional) – If True, show the plot immediately.
- biosppy.plotting.plot_biometrics(assessment=None, eer_idx=None, path=None, show=False)[source]
Create a summary plot of a biometrics test run.
- Parameters:
assessment (dict) – Classification assessment results.
eer_idx (int, optional) – Classifier reference index for the Equal Error Rate.
path (str, optional) – If provided, the plot will be saved to the specified file.
show (bool, optional) – If True, show the plot immediately.
- biosppy.plotting.plot_bvp(ts=None, raw=None, filtered=None, onsets=None, heart_rate_ts=None, heart_rate=None, path=None, show=False)[source]
Create a summary plot from the output of signals.bvp.bvp.
- Parameters:
ts (array) – Signal time axis reference (seconds).
raw (array) – Raw BVP signal.
filtered (array) – Filtered BVP signal.
onsets (array) – Indices of BVP pulse onsets.
heart_rate_ts (array) – Heart rate time axis reference (seconds).
heart_rate (array) – Instantaneous heart rate (bpm).
path (str, optional) – If provided, the plot will be saved to the specified file.
show (bool, optional) – If True, show the plot immediately.
- biosppy.plotting.plot_clustering(data=None, clusters=None, path=None, show=False)[source]
Create a summary plot of a data clustering.
- Parameters:
data (array) – An m by n array of m data samples in an n-dimensional space.
clusters (dict) – Dictionary with the sample indices (rows from data) for each cluster.
path (str, optional) – If provided, the plot will be saved to the specified file.
show (bool, optional) – If True, show the plot immediately.
- biosppy.plotting.plot_ecg(ts=None, raw=None, filtered=None, rpeaks=None, templates_ts=None, templates=None, heart_rate_ts=None, heart_rate=None, units=None, path=None, show=False)[source]
Create a summary plot from the output of signals.ecg.ecg.
- Parameters:
ts (array) – Signal time axis reference (seconds).
raw (array) – Raw ECG signal.
filtered (array) – Filtered ECG signal.
rpeaks (array) – R-peak location indices.
templates_ts (array) – Templates time axis reference (seconds).
templates (array) – Extracted heartbeat templates.
heart_rate_ts (array) – Heart rate time axis reference (seconds).
heart_rate (array) – Instantaneous heart rate (bpm).
units (str, optional) – Units of the vertical axis. If provided, the plot title will include the units information. Default is None.
path (str, optional) – If provided, the plot will be saved to the specified file.
show (bool, optional) – If True, show the plot immediately.
- biosppy.plotting.plot_eda(ts=None, raw=None, filtered=None, edr=None, edl=None, onsets=None, peaks=None, amplitudes=None, units=None, path=None, show=False)[source]
Create a summary plot from the output of signals.eda.eda.
- Parameters:
ts (array) – Signal time axis reference (seconds).
raw (array) – Raw EDA signal.
filtered (array) – Filtered EDA signal.
edr (array) – Electrodermal response signal.
edl (array) – Electrodermal level signal.
onsets (array) – Events onsets indices.
peaks (array) – Events peaks indices.
amplitudes (array) – Amplitudes location indices.
units (str, optional) – Units of the vertical axis. If provided, the plot title will include the units information. Default is None.
path (str, optional) – If provided, the plot will be saved to the specified file.
show (bool, optional) – If True, show the plot immediately.
- biosppy.plotting.plot_eeg(ts=None, raw=None, filtered=None, labels=None, features_ts=None, theta=None, alpha_low=None, alpha_high=None, beta=None, gamma=None, plf_pairs=None, plf=None, path=None, show=False)[source]
Create a summary plot from the output of signals.eeg.eeg.
- Parameters:
ts (array) – Signal time axis reference (seconds).
raw (array) – Raw EEG signal.
filtered (array) – Filtered EEG signal.
labels (list) – Channel labels.
features_ts (array) – Features time axis reference (seconds).
theta (array) – Average power in the 4 to 8 Hz frequency band; each column is one EEG channel.
alpha_low (array) – Average power in the 8 to 10 Hz frequency band; each column is one EEG channel.
alpha_high (array) – Average power in the 10 to 13 Hz frequency band; each column is one EEG channel.
beta (array) – Average power in the 13 to 25 Hz frequency band; each column is one EEG channel.
gamma (array) – Average power in the 25 to 40 Hz frequency band; each column is one EEG channel.
plf_pairs (list) – PLF pair indices.
plf (array) – PLF matrix; each column is a channel pair.
path (str, optional) – If provided, the plot will be saved to the specified file.
show (bool, optional) – If True, show the plot immediately.
- biosppy.plotting.plot_emg(ts=None, sampling_rate=None, raw=None, filtered=None, onsets=None, processed=None, units=None, path=None, show=False)[source]
Create a summary plot from the output of signals.emg.emg.
- Parameters:
ts (array) – Signal time axis reference (seconds).
sampling_rate (int, float) – Sampling frequency (Hz).
raw (array) – Raw EMG signal.
filtered (array) – Filtered EMG signal.
onsets (array) – Indices of EMG pulse onsets.
processed (array, optional) – Processed EMG signal according to the chosen onset detector.
units (str, optional) – Units of the vertical axis. If provided, the plot title will include the units information. Default is None.
path (str, optional) – If provided, the plot will be saved to the specified file.
show (bool, optional) – If True, show the plot immediately.
- biosppy.plotting.plot_filter(ftype='FIR', band='lowpass', order=None, frequency=None, sampling_rate=1000.0, log_xscale=True, path=None, show=True, **kwargs)[source]
Plot the frequency response of the filter specified with the given parameters.
- Parameters:
ftype (str) –
- Filter type:
Finite Impulse Response filter (‘FIR’);
Butterworth filter (‘butter’);
Chebyshev filters (‘cheby1’, ‘cheby2’);
Elliptic filter (‘ellip’);
Bessel filter (‘bessel’).
band (str) –
- Band type:
Low-pass filter (‘lowpass’);
High-pass filter (‘highpass’);
Band-pass filter (‘bandpass’);
Band-stop filter (‘bandstop’).
order (int) – Order of the filter.
frequency (int, float, list, array) –
- Cutoff frequencies; format depends on type of band:
’lowpass’ or ‘bandpass’: single frequency;
’bandpass’ or ‘bandstop’: pair of frequencies.
sampling_rate (int, float, optional) – Sampling frequency (Hz).
log_xscale (bool, optional) – Whether to use log scale for x-axis.
path (str, optional) – If provided, the plot will be saved to the specified file.
show (bool, optional) – If True, show the plot immediately.
**kwargs (dict, optional) – Additional keyword arguments are passed to the underlying scipy.signal function.
- biosppy.plotting.plot_hrv(rri, rri_trend=None, td_out=None, nl_out=None, fd_out=None, show=False)[source]
Create a summary plot of a HRV analysis.
- Parameters:
rri (array) – RR-intervals (ms).
rri_trend (array, optional) – RR-intervals trend (ms).
td_out (dict) – Output of signals.hrv.timedomain.
nl_out (dict) – Output of signals.hrv.nonlinear.
fd_out (dict) – Output of signals.hrv.frequencyomain.
show (bool, optional) – If True, show the plot immediately.
- biosppy.plotting.plot_hrv_fbands(frequencies=None, powers=None, fbands=None, method_name=None, legends=None, ax=None, show=False)[source]
Plots the power spectrum and highlights the defined frequency bands from the output of signals.hrv.compute_fbands.
- Parameters:
frequencies (array) – Frequency axis.
powers (array) – Power spectrum values for the frequency axis.
fbands (dict, optional) – Dictionary containing the limits of the frequency bands.
method_name (str, optional) – Method that was used to compute the power spectrum.
legends (dict, optional) – Additional legend elements.
ax (axis, optional) – Plot Axis to use.
show (bool, optional) – If True, show the plot immediately.
- biosppy.plotting.plot_hrv_hist(rri=None, bins=None, q_hist=None, hti=None, tinn=None, ax=None, show=False)[source]
Plots the RRI histogram with the corresponding geometrical HRV features from the output of signals.hrv.compute_geometrical.
- Parameters:
rri (array) – RR-intervals (ms).
bins (array) – Histogram bins.
q_hist (array) – Multilinear function fitted to the histogram.
hti (float) – HTI - HRV triangular index - Integral of the density of the RR interval histogram divided by its height.
tinn (float) – TINN - Baseline width of RR interval histogram (ms).
ax (axis, optional) – Plot Axis to use.
show (bool, optional) – If True, show the plot immediately.
- biosppy.plotting.plot_pcg(ts=None, raw=None, filtered=None, peaks=None, heart_sounds=None, heart_rate_ts=None, inst_heart_rate=None, units=None, path=None, show=False)[source]
Create a summary plot from the output of signals.pcg.pcg.
- Parameters:
ts (array) – Signal time axis reference (seconds).
raw (array) – Raw PCG signal.
filtered (array) – Filtered PCG signal.
peaks (array) – Peak location indices.
heart_sounds (array) – Classification of peaks as S1 or S2
heart_rate_ts (array) – Heart rate time axis reference (seconds).
inst_heart_rate (array) – Instantaneous heart rate (bpm).
units (str, optional) – Units of the vertical axis. If provided, the plot title will include the units information. Default is None.
path (str, optional) – If provided, the plot will be saved to the specified file.
show (bool, optional) – If True, show the plot immediately.
- biosppy.plotting.plot_poincare(rri=None, s=None, sd1=None, sd2=None, legends=None, ax=None, show=False)[source]
Plot a Poincaré plot of a series of RR intervals (RRI[i+1] vs. RRI[i]) from the output of signals.hrv.compute_poincare.
- Parameters:
rri (array) – RR-intervals (ms).
s (float) – S - Area of the ellipse of the Poincaré plot (ms^2).
sd1 (float) – SD1 - Poincaré plot standard deviation perpendicular to the identity line (ms).
sd2 (float) – SD2 - Poincaré plot standard deviation along the identity line (ms).
legends (dict, optional) – Dictionary of features to add to the plot legend.
ax (axis, optional) – Plot Axis to use.
show (bool, optional) – If True, show the plot immediately.
- biosppy.plotting.plot_ppg(ts=None, raw=None, filtered=None, peaks=None, templates_ts=None, templates=None, heart_rate_ts=None, heart_rate=None, units=None, path=None, show=False)[source]
Create a summary plot from the output of signals.ppg.ppg.
- Parameters:
ts (array) – Signal time axis reference (seconds).
raw (array) – Raw PPG signal.
filtered (array) – Filtered PPG signal.
peaks (array) – Indices of PPG pulse peaks.
templates_ts (array) – Templates time axis reference (seconds).
templates (array) – Extracted PPG templates.
heart_rate_ts (array) – Heart rate time axis reference (seconds).
heart_rate (array) – Instantaneous heart rate (bpm).
units (str, optional) – Units of the vertical axis. If provided, the plot title will include the units information. Default is None.
path (str, optional) – If provided, the plot will be saved to the specified file.
show (bool, optional) – If True, show the plot immediately.
- biosppy.plotting.plot_resp(ts=None, raw=None, filtered=None, zeros=None, resp_rate_ts=None, resp_rate=None, units=None, path=None, show=False)[source]
Create a summary plot from the output of signals.ppg.ppg.
- Parameters:
ts (array) – Signal time axis reference (seconds).
raw (array) – Raw Resp signal.
filtered (array) – Filtered Resp signal.
zeros (array) – Indices of Respiration zero crossings.
resp_rate_ts (array) – Respiration rate time axis reference (seconds).
resp_rate (array) – Instantaneous respiration rate (Hz).
units (str, optional) – Units of the vertical axis. If provided, the plot title will include the units information. Default is None.
path (str, optional) – If provided, the plot will be saved to the specified file.
show (bool, optional) – If True, show the plot immediately.
- biosppy.plotting.plot_rri(rri, rri_trend=None, legends=None, ax=None, show=False)[source]
Plot a series of RR intervals.
- Parameters:
rri (array) – RR-intervals (ms).
rri_trend (array, optional) – RR-intervals trend (ms).
legends (dict, optional) – Dictionary of features to add to the plot legend.
ax (axis, optional) – Plot Axis to use.
show (bool, optional) – If True, show the plot immediately.
- biosppy.plotting.plot_spectrum(signal=None, sampling_rate=1000.0, path=None, show=True)[source]
Plot the power spectrum of a signal (one-sided).
- Parameters:
signal (array) – Input signal.
sampling_rate (int, float, optional) – Sampling frequency (Hz).
path (str, optional) – If provided, the plot will be saved to the specified file.
show (bool, optional) – If True, show the plot immediately.
biosppy.quality
This provides functions to assess the quality of several biosignals.
- copyright:
2015-2023 by Instituto de Telecomunicacoes
- license:
BSD 3-clause, see LICENSE for more details.
- biosppy.quality.cSQI(rpeaks=None, verbose=1)[source]
For the ECG signal Calculate the Coefficient of Variation of RR Intervals (cSQI). :param rpeaks: Array containing R-peak locations. Should be filtered? How many seconds are adequate? :type rpeaks: array-like :param verbose: If 1, a commentary is printed regarding the quality of the signal and details of the function. Default is 1. :type verbose: int
- Returns:
cSQI (float) – Coefficient of Variation of RR Intervals. cSQI - best near 0
References
[Zhao18]Zhao, Z., & Zhang, Y. (2018).
SQI quality evaluation mechanism of single-lead ECG signal based on simple heuristic fusion and fuzzy comprehensive evaluation. Frontiers in Physiology, 9, 727.
- biosppy.quality.ecg_sqi_level3(segment, sampling_rate, threshold, bit)[source]
Compute the quality index for one ECG segment. The segment should have 10 seconds.
- Parameters:
segment (array) – Input signal to test.
sampling_rate (int) – Sampling frequency (Hz).
threshold (float) – Threshold for the correlation coefficient.
bit (int) – Number of bits of the ADC.? Resolution bits, for the BITalino is 10 bits.
- Returns:
quality (string) – Signal Quality Index ranging between 0 (LQ), 0.5 (MQ) and 1.0 (HQ).
- biosppy.quality.eda_sqi_bottcher(x=None, sampling_rate=None, verbose=1)[source]
Suggested by Böttcher et al. Scientific Reports, 2022, for wearable wrist EDA. This is given by a binary score 0/1 defined by the following rules: - mean of the segment of 2 seconds should be > 0.05 - rate of amplitude change (given by racSQI) should be < 0.2 This score is calculated for each 2 seconds window of the segment. The average of the scores is the final SQI. This method was designed for a segment of 60s
- Parameters:
x (array) – Input signal to test.
sampling_rate (int) – Sampling frequency (Hz).
verbose (int) – If 1, a commentary is printed regarding the quality of the signal and details of the function. Default is 1.
- Returns:
quality_score (string) – Signal Quality Index.
- biosppy.quality.hosSQI(signal=None, quantitative=False, verbose=1)[source]
For the ECG signal. Calculate the Higher-order-statistics-SQI (hosSQI). :param signal: ECG signal. Should be filtered? How many seconds are adequate? :type signal: array-like :param verbose: If True, a warning message is printed. Default is True. :type verbose: bool
- Returns:
hosSQI (float) – Higher-order-statistics-SQI. hosSQI - best near 1
References
[Nardelli20]Nardelli, M., Lanata, A., Valenza, G., Felici, M., Baragli, P., & Scilingo, E.P. (2020).
A tool for the real-time evaluation of ECG signal quality and activity: Application to submaximal treadmill test in horses. Biomedical Signal Processing and Control, 56, 101666. doi: 10.1016/j.bspc.2019.101666. .. [Rahman22] Rahman, Md. Saifur, Karmakar, Chandan, Natgunanathan, Iynkaran, Yearwood, John, & Palaniswami, Marimuthu. (2022). Robustness of electrocardiogram signal quality indices. Journal of The Royal Society Interface, 19. doi: 10.1098/rsif.2022.0012.
- biosppy.quality.quality_ecg(segment, methods=['Level3'], sampling_rate=None, fisher=True, f_thr=0.01, threshold=0.9, bit=0, nseg=1024, num_spectrum=[5, 20], dem_spectrum=None, mode_fsqi='simple', verbose=1)[source]
Compute the quality index for one ECG segment.
- Parameters:
segment (array) – Input signal to test.
method (string) – Method to assess quality. One of the following: ‘Level3’, ‘pSQI’, ‘kSQI’, ‘fSQI’.
sampling_rate (int) – Sampling frequency (Hz).
threshold (float) – Threshold for the correlation coefficient.
bit (int) – Number of bits of the ADC. Resolution bits, for the BITalino is 10 bits.
verbose (int) – If 1, a commentary is printed regarding the quality of the signal and details of the function. Default is 1.
- Returns:
args (tuple) – Tuple containing the quality index for each method.
names (tuple) – Tuple containing the name of each method.
- biosppy.quality.quality_eda(x=None, methods=['bottcher'], sampling_rate=None, verbose=1)[source]
Compute the quality index for one EDA segment.
- Parameters:
x (array) – Input signal to test.
methods (list) – Method to assess quality. One or more of the following: ‘bottcher’.
sampling_rate (int) – Sampling frequency (Hz).
verbose (int) – If 1, a commentary is printed regarding the quality of the signal and details of the function. Default is 1.
- Returns:
args (tuple) – Tuple containing the quality index for each method.
names (tuple) – Tuple containing the name of each method.
biosppy.stats
This module provides statistical functions and related tools.
- copyright:
2015-2023 by Instituto de Telecomunicacoes
- license:
BSD 3-clause, see LICENSE for more details.
- biosppy.stats.diff_stats(signal=None, stats_only=True)[source]
Compute statistical features from the first signal differences, second signal differences and absolute signal differences.
- Parameters:
signal (array) – Input signal.
stats_only (bool, optional) – Whether to output only statistical features. Default is True.
- Returns:
{diff} (array) – Difference signal. {diff} can be ‘diff’, ‘diff2’ or ‘abs_diff’.
{diff}_mean (float) – Mean of the difference signal.
{diff}_median (float) – Median of the difference signal.
{diff}_min (float) – Minimum of the difference signal.
{diff}_max (float) – Maximum of the difference signal.
{diff}_max_amp (float) – Maximum amplitude of the difference signal.
{diff}_range (float) – Range of the difference signal.
{diff}_var (float) – Variance of the difference signal.
{diff}_std (float) – Standard deviation of the difference signal.
{diff}_sum (float) – Sum of the difference signal.
- biosppy.stats.histogram(signal=None, bins=5, normalize=True)[source]
Compute histogram of the input signal.
- Parameters:
signal (array) – Input signal.
bins (int, optional) – Number of histogram bins. Default is 5.
normalize (bool, optional) – Whether to normalize the histogram counts. Default is True.
- Returns:
hist{bin}_bins (float) – Number of counts of the bin. If normalize is True, the counts are normalized.
- biosppy.stats.linear_regression(x=None, y=None, show=True)[source]
Plot the linear regression between two signals and get the equation coefficients.
The linear regression uses the least squares method.
- Parameters:
x (array) – First input signal.
y (array) – Second input signal.
show (bool) – If True, show the plot.
- Returns:
coeffs (array) – Linear regression coefficients: [m, b].
- Raises:
ValueError – If the input signals do not have the same length.
- biosppy.stats.paired_test(x=None, y=None)[source]
Perform the Student’s paired t-test on the arrays x and y. This is a two-sided test for the null hypothesis that 2 related or repeated samples have identical average (expected) values.
- Parameters:
x (array) – First input signal.
y (array) – Second input signal.
- Returns:
statistic (float) – t-statistic. The t-statistic is used in a t-test to determine if you should support or reject the null hypothesis.
pvalue (float) – Two-sided p-value.
- Raises:
ValueError – If the input signals do not have the same length.
- biosppy.stats.pearson_correlation(x=None, y=None)[source]
Compute the Pearson Correlation Coefficient between two signals.
The coefficient is given by:
![r_{xy} = \frac{E[(X - \mu_X) (Y - \mu_Y)]}{\sigma_X \sigma_Y}](_images/math/25984ac0b99af0b57d7d197c63928096548adcb5.png)
- Parameters:
x (array) – First input signal.
y (array) – Second input signal.
- Returns:
r (float) – Pearson correlation coefficient, ranging between -1 and +1.
pvalue (float) – Two-tailed p-value. The p-value roughly indicates the probability of an uncorrelated system producing datasets that have a Pearson correlation at least as extreme as the one computed from these datasets.
- Raises:
ValueError – If the input signals do not have the same length.
- biosppy.stats.quartiles(signal=None)[source]
Compute quartile features of the signal.
- Parameters:
signal (array) – Input signal.
- Returns:
q1 (float) – First quartile.
q2 (float) – Second quartile, also known as median.
q3 (float) – Third quartile.
iqr (float) – Interquartile range.
midhinge (float) – Midhinge.
trimean (float) – Trimean.
- biosppy.stats.unpaired_test(x=None, y=None)[source]
Perform the Student’s unpaired t-test on the arrays x and y. This is a two-sided test for the null hypothesis that 2 independent samples have identical average (expected) values. This test assumes that the populations have identical variances by default.
- Parameters:
x (array) – First input signal.
y (array) – Second input signal.
- Returns:
statistic (float) – t-statistic. The t-statistic is used in a t-test to determine if you should support or reject the null hypothesis.
pvalue (float) – Two-sided p-value.
- Raises:
ValueError – If the input signals do not have the same length.
biosppy.storage
This module provides several data storage methods.
- copyright:
2015-2018 by Instituto de Telecomunicacoes
- license:
BSD 3-clause, see LICENSE for more details.
- class biosppy.storage.HDF(path=None, mode='a')[source]
Bases:
objectWrapper class to operate on BioSPPy HDF5 files.
- Parameters:
path (str) – Path to the HDF5 file.
mode (str, optional) –
File mode; one of:
’a’: read/write, creates file if it does not exist;
’r+’: read/write, file must exist;
’r’: read only, file must exist;
’w’: create file, truncate if it already exists;
’w-’: create file, fails if it already esists.
- add_event(ts=None, values=None, mdata=None, group='', name=None, compress=False)[source]
Add an event to the file.
- Parameters:
ts (array) – Array of time stamps.
values (array, optional) – Array with data for each time stamp.
mdata (dict, optional) – Event metadata.
group (str, optional) – Destination event group.
name (str, optional) – Name of the dataset to create.
compress (bool, optional) – If True, the data will be compressed with gzip.
- Returns:
group (str) – Destination group.
name (str) – Name of the created event dataset.
- add_signal(signal=None, mdata=None, group='', name=None, compress=False)[source]
Add a signal to the file.
- Parameters:
signal (array) – Signal to add.
mdata (dict, optional) – Signal metadata.
group (str, optional) – Destination signal group.
name (str, optional) – Name of the dataset to create.
compress (bool, optional) – If True, the signal will be compressed with gzip.
- Returns:
group (str) – Destination group.
name (str) – Name of the created signal dataset.
- del_event(group='', name=None)[source]
Delete an event from the file.
- Parameters:
group (str, optional) – Event group.
name (str) – Name of the event dataset.
- del_event_group(group='')[source]
Delete all events in a file group.
- Parameters:
str (group) – Event group.
optional – Event group.
- del_signal(group='', name=None)[source]
Delete a signal from the file.
- Parameters:
group (str, optional) – Signal group.
name (str) – Name of the dataset.
- del_signal_group(group='')[source]
Delete all signals in a file group.
- Parameters:
group (str, optional) – Signal group.
- get_event(group='', name=None)[source]
Retrieve an event from the file.
- Parameters:
group (str, optional) – Event group.
name (str) – Name of the event dataset.
- Returns:
ts (array) – Array of time stamps.
values (array) – Array with data for each time stamp.
mdata (dict) – Event metadata.
Notes
Loads the entire event data into memory.
- get_signal(group='', name=None)[source]
Retrieve a signal from the file.
- Parameters:
group (str, optional) – Signal group.
name (str) – Name of the signal dataset.
- Returns:
signal (array) – Retrieved signal.
mdata (dict) – Signal metadata.
Notes
Loads the entire signal data into memory.
- biosppy.storage.alloc_h5(path)[source]
Prepare an HDF5 file.
- Parameters:
path (str) – Path to file.
- biosppy.storage.deserialize(path)[source]
Deserialize data from a file using sklearn’s joblib.
- Parameters:
path (str) – Source path.
- Returns:
data (object) – Deserialized object.
- biosppy.storage.dumpJSON(data, path)[source]
Save JSON data to a file.
- Parameters:
data (dict) – The JSON data to dump.
path (str) – Destination path.
- biosppy.storage.loadJSON(path)[source]
Load JSON data from a file.
- Parameters:
path (str) – Source path.
- Returns:
data (dict) – The loaded JSON data.
- biosppy.storage.load_h5(path, label)[source]
Load data from an HDF5 file.
- Parameters:
path (str) – Path to file.
label (hashable) – Data label.
- Returns:
data (array) – Loaded data.
- biosppy.storage.load_txt(path)[source]
Load data from a text file.
- Parameters:
path (str) – Path to file.
- Returns:
data (array) – Loaded data.
mdata (dict) – Metadata.
- biosppy.storage.pack_zip(files, path, recursive=True, forceExt=True)[source]
Pack files into a zip archive.
- Parameters:
files (iterable) – List of files or directories to pack.
path (str) – Destination path.
recursive (bool, optional) – If True, sub-directories and sub-folders are also written to the archive.
forceExt (bool, optional) – Append default extension.
- Returns:
zip_path (str) – Full path to created zip archive.
- biosppy.storage.serialize(data, path, compress=3)[source]
Serialize data and save to a file using sklearn’s joblib.
- Parameters:
data (object) – Object to serialize.
path (str) – Destination path.
compress (int, optional) – Compression level; from 0 to 9 (highest compression).
- biosppy.storage.store_h5(path, label, data)[source]
Store data to HDF5 file.
- Parameters:
path (str) – Path to file.
label (hashable) – Data label.
data (array) – Data to store.
- biosppy.storage.store_txt(path, data, sampling_rate=1000.0, resolution=None, date=None, labels=None, precision=6)[source]
Store data to a simple text file.
- Parameters:
path (str) – Path to file.
data (array) – Data to store (up to 2 dimensions).
sampling_rate (int, float, optional) – Sampling frequency (Hz).
resolution (int, optional) – Sampling resolution.
date (datetime, str, optional) – Datetime object, or an ISO 8601 formatted date-time string.
labels (list, optional) – Labels for each column of data.
precision (int, optional) – Precision for string conversion.
- Raises:
ValueError – If the number of data dimensions is greater than 2.
ValueError – If the number of labels is inconsistent with the data.
- biosppy.storage.unpack_zip(zip_path, path)[source]
Unpack a zip archive.
- Parameters:
zip_path (str) – Path to zip archive.
path (str) – Destination path (directory).
- biosppy.storage.zip_write(fid, files, recursive=True, root=None)[source]
Write files to zip archive.
- Parameters:
fid (file-like object) – The zip file to write into.
files (iterable) – List of files or directories to pack.
recursive (bool, optional) – If True, sub-directories and sub-folders are also written to the archive.
root (str, optional) – Relative folder path.
Notes
Ignores non-existent files and directories.
biosppy.timing
This module provides simple methods to measure computation times.
- copyright:
2015-2018 by Instituto de Telecomunicacoes
- license:
BSD 3-clause, see LICENSE for more details.
- biosppy.timing.clear(name=None)[source]
Clear the clock.
- Parameters:
name (str, optional) – Name of the clock; if None, uses the default name.
- biosppy.timing.tac(name=None)[source]
Stop the clock.
- Parameters:
name (str, optional) – Name of the clock; if None, uses the default name.
- Returns:
delta (float) – Elapsed time, in seconds.
- Raises:
KeyError if the name of the clock is unknown. –
- biosppy.timing.tic(name=None)[source]
Start the clock.
- Parameters:
name (str, optional) – Name of the clock; if None, uses the default name.
biosppy.utils
This module provides several frequently used functions and hacks.
- copyright:
2015-2018 by Instituto de Telecomunicacoes
- license:
BSD 3-clause, see LICENSE for more details.
- class biosppy.utils.ReturnTuple(values, names=None)[source]
Bases:
tupleA named tuple to use as a hybrid tuple-dict return object.
- Parameters:
values (iterable) – Return values.
names (iterable, optional) – Names for return values.
- Raises:
ValueError – If the number of values differs from the number of names.
ValueError – If any of the items in names: * contain non-alphanumeric characters; * are Python keywords; * start with a number; * are duplicates.
- append(new_values: (<class 'int'>, <class 'float'>, <class 'complex'>, <class 'str'>, typing.Collection, <Mock id='140169201848384'>), new_keys=None)[source]
Returns a new ReturnTuple with the new values and keys appended to the original object.
- Parameters:
new_values (int, float, list, tuple, dict, array) – Values to append. If given a dict and new_keys is None, the correspondent keys and values will be used.
new_keys (str, list, tuple, optional) – Keys to append.
- Returns:
object (ReturnTuple) – A ReturnTuple with the values and keys appended.
- as_dict()[source]
Convert to an ordered dictionary.
- Returns:
out (OrderedDict) – An OrderedDict representing the return values.
- delete(key)[source]
Returns a ReturnTuple without the specified key.
- Parameters:
key (str) – ReturnTuple key to be deleted.
- Returns:
object (ReturnTuple) – The ReturnTuple with the key removed.
- join(new_tuple)[source]
Returns a ReturnTuple with the new ReturnTuple appended.
- Parameters:
new_tuple (ReturnTuple) – ReturnTuple to be joined.
- Returns:
object (ReturnTuple) – The joined ReturnTuple.
- biosppy.utils.fileparts(path)[source]
split a file path into its directory, name, and extension.
- Parameters:
path (str) – Input file path.
- Returns:
dirname (str) – File directory.
fname (str) – File name.
ext (str) – File extension.
Notes
Removes the dot (‘.’) from the extension.
- biosppy.utils.fullfile(*args)[source]
Join one or more file path components, assuming the last is the extension.
- Parameters:
*args (list, optional) – Components to concatenate.
- Returns:
fpath (str) – The concatenated file path.
- biosppy.utils.highestAveragesAllocator(votes, k, divisor='dHondt', check=False)[source]
Allocate k seats proportionally using the Highest Averages Method.
- Parameters:
votes (list) – Number of votes for each class/party/cardinal.
k (int) – Total number o seats to allocate.
divisor (str, optional) – Divisor method; one of ‘dHondt’, ‘Huntington-Hill’, ‘Sainte-Lague’, ‘Imperiali’, or ‘Danish’.
check (bool, optional) – If True, limits the number of seats to the total number of votes.
- Returns:
seats (list) – Number of seats for each class/party/cardinal.
- biosppy.utils.normpath(path)[source]
Normalize a path.
- Parameters:
path (str) – The path to normalize.
- Returns:
npath (str) – The normalized path.
- biosppy.utils.random_fraction(indx, fraction, sort=True)[source]
Select a random fraction of an input list of elements.
- Parameters:
indx (list, array) – Elements to partition.
fraction (int, float) – Fraction to select.
sort (bool, optional) – If True, output lists will be sorted.
- Returns:
use (list, array) – Selected elements.
unuse (list, array) – Remaining elements.
- biosppy.utils.remainderAllocator(votes, k, reverse=True, check=False)[source]
Allocate k seats proportionally using the Remainder Method.
Also known as Hare-Niemeyer Method. Uses the Hare quota.
- Parameters:
votes (list) – Number of votes for each class/party/cardinal.
k (int) – Total number o seats to allocate.
reverse (bool, optional) – If True, allocates remaining seats largest quota first.
check (bool, optional) – If True, limits the number of seats to the total number of votes.
- Returns:
seats (list) – Number of seats for each class/party/cardinal.
- biosppy.utils.walktree(top=None, spec=None)[source]
Iterator to recursively descend a directory and return all files matching the spec.
- Parameters:
top (str, optional) – Starting directory; if None, defaults to the current working directoty.
spec (str, optional) – Regular expression to match the desired files; if None, matches all files; typical patterns: * r’.txt$’ - matches files with ‘.txt’ extension; * r’^File_’ - matches files starting with ‘File_’ * r’^File_.+.txt$’ - matches files starting with ‘File_’ and ending with the ‘.txt’ extension.
- Yields:
fpath (str) – Absolute file path.
Notes
Partial matches are also selected.